
Als we het over neurologische aandoeningen hebben, zijn er maar weinig aandoeningen die zo ingewikkeld en verwoestend zijn als Amyotrofische Lateraal Sclerose, beter bekend als ALS. Deze raadselachtige ziekte, vaak synoniem met de ziekte van Lou Gehrig ter ere van de gevierde honkbalspeler wiens carrière er tragisch door werd beïnvloed, is een ingewikkeld raadsel dat de aandacht blijft trekken van medische onderzoekers, clinici en het grote publiek.
Hoewel ALS misschien niet dezelfde wijdverspreide erkenning geniet als bepaalde andere neurologische aandoeningen, is de impact op zowel de direct getroffenen als hun dierbaren diepgaand en gaat vaak gepaard met hartverscheurende emoties. Dit artikel, getiteld "Wat is Amyotrofische Lateraal Sclerose en de gevolgen", gaat in op de kern van deze tragische aandoening.
Helaas heeft het recente verlies van Bryan Randall, een fotograaf en oude partner van Sandra Bullock, ALS opnieuw onder de aandacht gebracht. Bryan's overlijden op 57-jarige leeftijd benadrukte het meedogenloze karakter van de ziekte. Helaas bestaat er geen genezing voor ALS en na de diagnose hebben mensen meestal een gemiddelde levensverwachting van 2 tot 5 jaar.
Wat is Amyotrofische Lateraal Sclerose?
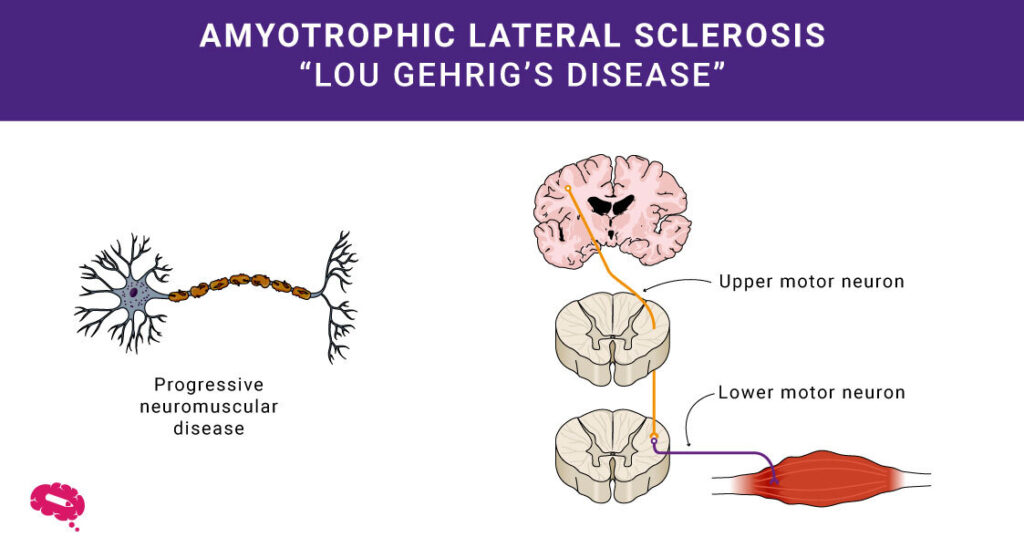
De medische wetenschap legt vaak complexe mysteries bloot op het gebied van de menselijke gezondheid. Eén daarvan is Amyotrofische Lateraal Sclerose, of ALS. Je kent het misschien als de ziekte van Lou Gehrig. Deze aandoening tast de motorneuronen in de hersenen en het ruggenmerg aan, die de menselijke spieren aansturen. Bij ALS gaan deze neuronen kapot, waardoor het moeilijk wordt om spieren te bewegen.
Definitie van ALS
ALS, of Amyotrofische Lateraal Sclerose, is een aandoening die de werking van de menselijke spieren beïnvloedt. Hierdoor worden de menselijke spieren na verloop van tijd zwakker, wat kan leiden tot problemen met bewegen en het uitvoeren van alledaagse taken.
Korte geschiedenis en achtergrond
De geschiedenis van Amyotrofische Lateraal Sclerose (ALS) is een verhaal van ontdekkingen dat meer dan een eeuw omspant. Het begon in 1869 toen de Franse neuroloog Jean-Martin Charcot de ziekte voor het eerst identificeerde en beschreef. ALS kreeg echter meer bekendheid in 1939 toen de legendarische honkbalspeler Lou Gehrig de diagnose kreeg, wat leidde tot de volksnaam "de ziekte van Lou Gehrig".
Vooruitgang in de jaren 1960 en 1970 bracht de kern van de ziekte aan het licht en onthulde de progressieve degeneratie van motorische neuronen als onderliggend mechanisme. In de jaren 1990 brachten genetische doorbraken verbanden aan het licht tussen ALS en genen zoals SOD1 en TDP-43, waardoor ons begrip van de genetische componenten werd verdiept.
De 21e eeuw werd gekenmerkt door belangrijke mijlpalen, zoals de virale Ice Bucket Challenge die zowel bewustzijn als middelen voor ALS-onderzoek opwekte. In 2016 keurde de FDA het eerste ALS-specifieke medicijn goed, edaravone (Radicava), een nieuwe aanpak om de ziekteprogressie te vertragen.
Terwijl we het jaar 2020 doorkruisen, gaat het onderzoek onvermoeibaar door. We onderzoeken de mogelijkheden voor behandeling, de genetische complexiteit van ALS en de onderliggende oorzaken. Deze reis onderstreept onze toewijding aan het ontrafelen van de complexiteit van ALS en het verlichten van de impact op de getroffenen.
Het belang van ALS begrijpen
Inzicht in ALS speelt een belangrijke rol voor verschillende individuen en gaat verder dan medische experts en omvat de hele maatschappij. Het belang van dit begrip komt voort uit verschillende belangrijke redenen:
Ten eerste maakt ALS geen onderscheid op basis van demografie, waardoor een wijdverspreid begrip van cruciaal belang is. Een grotere bekendheid met de ziekte stelt medische professionals in staat om de ziekte in een vroeg stadium te herkennen, zodat er tijdig kan worden ingegrepen en zorg kan worden verleend aan ALS-patiënten.
Ten tweede biedt een beter begrip van ALS een bron van hoop voor gezinnen die met deze uitdagingen geconfronteerd worden. Duidelijke inzichten in het verloop van de ziekte en de beschikbare behandelingen stellen gezinnen in staat om vooruit te plannen, hun leven aan te passen en weloverwogen keuzes te maken.
Bovendien winnen de onderzoeksinspanningen aan kracht door een beter begrip. Een goed begrip van de ingewikkelde aard van ALS versnelt de zoektocht naar mogelijke therapieën en interventies. Met een beter begrip kunnen middelen efficiënter worden ingezet voor effectieve oplossingen.
Uiteindelijk zorgt het begrijpen van ALS voor een collectief front. Families, vrienden, zorgverleners, onderzoekers en de bredere gemeenschap verenigen zich om deze uitdaging aan te gaan. Door dit alles te begrijpen, ontstaat er een krachtige beweging tegen ALS die een toekomst voor ogen heeft waarin ALS synoniem is met veerkracht, solidariteit en vooruitgang.
De ziekte begrijpen
Amyotrofische Lateraal Sclerose (ALS) staat bekend als een complexe ziekte. Hier zijn een aantal cruciale details om een licht op te werpen:
Symptomen en vroege symptomen
ALS kan alleen in een vroeg stadium worden ontdekt door de kenmerkende symptomen te identificeren. De eerste aanwijzingen zijn vaak verzwakte spieren, vooral in de ledematen. Andere mogelijke symptomen zijn spraakstoornissen, spiertrekkingen en vermoeidheid.
Oorzaken en risicofactoren
De exacte oorzaak van ALS is nog niet volledig bekend, maar men denkt dat het een gevolg is van een combinatie van genetische en omgevingsfactoren. Hier zijn enkele bekende oorzaken en risicofactoren van ALS:
- Genetische factoren: Een klein percentage (ongeveer 5-10%) van de ALS-gevallen is erfelijk, wat betekent dat ze worden veroorzaakt door mutaties in specifieke genen. Het meest bekende gen dat in verband wordt gebracht met familiaire ALS is de C9orf72 genmutatie. Mutaties in andere genen zoals SOD1, TARDBP en FUS zijn ook in verband gebracht met ALS. Deze genetische mutaties kunnen leiden tot de ophoping van abnormale eiwitten in neuronen, wat schade en celdood veroorzaakt.
- Omgevingsfactoren: Hoewel genetische factoren een belangrijke rol spelen in familiale gevallen van ALS, wordt ook aangenomen dat omgevingsfactoren bijdragen aan de ontwikkeling van de ziekte. Blootstelling aan bepaalde toxines en chemicaliën, zoals zware metalen, pesticiden en industriële vervuilende stoffen, wordt voorgesteld als mogelijke risicofactoren.
- Leeftijd: ALS ontwikkelt zich meestal bij volwassenen tussen 40 en 70 jaar, met een gemiddelde leeftijd tussen 55 en 60 jaar. Het kan echter op elke leeftijd optreden.
- Geslacht: ALS komt iets vaker voor bij mannen dan bij vrouwen. De redenen voor dit verschil tussen mannen en vrouwen worden niet volledig begrepen.
- Ras en etniciteit: ALS komt voor in alle raciale en etnische groepen, maar het lijkt vaker voor te komen bij blanken in vergelijking met andere raciale groepen.
- Roken: Er zijn aanwijzingen dat roken het risico op het ontwikkelen van ALS verhoogt. Roken wordt in verband gebracht met oxidatieve stress en ontstekingen, waarvan wordt aangenomen dat ze een rol spelen bij de progressie van de ziekte.
- Lichamelijke activiteit: Er zijn aanwijzingen dat personen met een geschiedenis van veel lichaamsbeweging een licht verhoogd risico op ALS hebben. De relatie tussen lichaamsbeweging en het risico op ALS is echter niet volledig duidelijk en moet verder worden onderzocht.
- Hoofdtrauma: Er zijn suggesties dat een voorgeschiedenis van traumatisch hoofdletsel in verband kan worden gebracht met een licht verhoogd risico op ALS. Het verband tussen hoofdtrauma en ALS wordt echter nog steeds onderzocht.
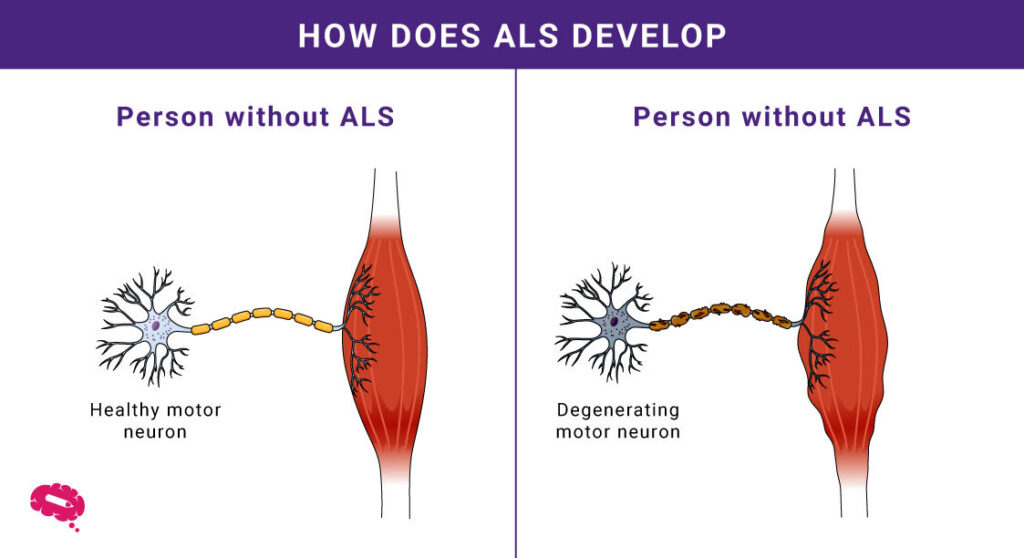
Hoe ALS het lichaam beïnvloedt
- Degeneratie van motorneuronen: ALS richt zich op motorische neuronen die spierzwakte en controleverlies veroorzaken.
- Spierzwakte: Geleidelijke degeneratie leidt tot problemen met bewegen en alledaagse taken.
- Spraak- en slikproblemen: Spreek- en slikspieren worden aangetast, wat slissen en moeite met eten veroorzaakt.
- Ademhalingsproblemen: De ademhalingsspieren verzwakken, wat leidt tot ademhalingsmoeilijkheden.
- Progressieve verlamming: Na verloop van tijd tast toenemende verlamming de mobiliteit en onafhankelijkheid aan.
- Cognitieve veranderingen: Sommigen ervaren veranderingen in het geheugen en de besluitvorming, vooral bij ALS met frontotemporale dementie.
- Zintuiglijke functie intact: Gevoelens zoals aanraken en zien blijven meestal onaangetast.
- Levensbedreigende complicaties: Ademhalingsfalen in een gevorderd stadium kan levensbedreigend zijn.
Diagnose en testen
De diagnose ALS is een uitgebreid proces dat klinische evaluatie, beoordeling van de medische voorgeschiedenis, neurologisch onderzoek en de uitsluiting van alternatieve aandoeningen omvat. De diagnose berust op een reeks onderzoeken, waaronder elektromyografie (EMG), zenuwgeleidingsonderzoek (NCS) en beeldvorming, die samen de zenuw- en spierfunctie evalueren. Bij ALS is er niet één definitieve test; de diagnose berust eerder op een combinatie van klinische observaties en resultaten van verschillende onderzoeken.
Diagnostische procedures
- Controle van de medische voorgeschiedenis om inzicht te krijgen in de symptomen en hun verloop.
- Een neurologische beoordeling uitvoeren om spierkracht, reflexen en coördinatie te evalueren.
- Elektromyografie (EMG) meet spieractiviteit en spoort zenuwproblemen op.
- Zenuwgeleidingsonderzoek (NCS) beoordeelt de zenuwfunctie en -respons.
Uitdagingen in diagnose
- ALS symptomen overlappen met andere aandoeningen, wat kan leiden tot een verkeerde diagnose of een vertraagde diagnose.
- Een gebrek aan specifieke biomarkers kan een vroege en nauwkeurige diagnose in de weg staan.
- Progressie en symptoomvariatie maken de diagnose moeilijk.
ALS onderscheiden van andere ziekten
In tegenstelling tot multiple sclerose (MS), dat vooral de myelineschede van het centrale zenuwstelsel aantast, richt ALS zich op de motorneuronen die verantwoordelijk zijn voor de vrijwillige controle over de spieren.
Hoewel primaire laterale sclerose (PLS) bepaalde kenmerken gemeen heeft met ALS, zijn er voornamelijk bovenste motorneuronen bij betrokken, in tegenstelling tot de gecombineerde betrokkenheid van bovenste en onderste motorneuronen bij ALS.
Daarnaast is het van cruciaal belang om ALS te onderscheiden van spinale musculaire atrofie (SMA) vanwege hun gedeelde impact op motorische neuronen; SMA tast echter voornamelijk de lagere motorische neuronen aan, in tegenstelling tot ALS. Het vermogen om ALS van deze ziekten te onderscheiden is essentieel voor een nauwkeurige diagnose en behandelingsstrategieën op maat.
Behandeling en beheer
De behandeling en het beheer van ALS omvatten een veelzijdige aanpak. De beschikbare behandelingsmogelijkheden zijn erop gericht de symptomen te verlichten, de ziekteprogressie te vertragen en de levenskwaliteit van ALS-patiënten te verbeteren.
Beschikbare behandelingsopties
- De behandeling van ALS is er vooral op gericht om de symptomen aan te pakken, de levenskwaliteit te verbeteren en de progressie van de ziekte te vertragen.
- Medicijnen zoals riluzole en edaravone zijn goedgekeurd om mogelijk de overleving te verlengen en progressie te vertragen.
- Baclofen en andere medicijnen kunnen de spierspasticiteit helpen beheersen, terwijl medicijnen zoals anticholinergica de overmatige speekselproductie aanpakken.
- Hulpmiddelen, zoals niet-invasieve beademingsapparatuur en voedingssonde, helpen bij de ademhaling en voeding als de spierfunctie afneemt.
Symptomen en levenskwaliteit beheren
- Fysiotherapie en ergotherapie helpen bij het behouden van spierkracht, mobiliteit en onafhankelijkheid.
- Logopedie helpt bij communicatie- en slikproblemen.
- Ondersteunende therapieën, zoals voedingsadviezen, psychologische begeleiding en steungroepen, helpen bij het aanpakken van emotionele en praktische problemen.
- Palliatieve zorg en hospicezorg verbeteren het comfort en de levenskwaliteit in gevorderde stadia.
Lopend onderzoek en toekomstige therapieën
- Onderzoekers blijven de onderliggende oorzaken van ALS en mogelijke behandelingsdoelen onderzoeken.
- Gentherapie is gericht op het corrigeren van genetische mutaties die verantwoordelijk zijn voor familiaire ALS.
- Stamcelonderzoek onderzoekt het gebruik van stamcellen om beschadigde motorische neuronen te vervangen.
- Innovatieve benaderingen zijn onder andere het aanpakken van abnormale eiwitophoping en ontsteking in door ALS aangetaste neuronen.
- Het ontwikkelen van biomarkers kan helpen bij een vroege diagnose en het volgen van de progressie van de ziekte.
- Er wordt nog steeds gezocht naar nieuwe therapeutische middelen en strategieën om ALS effectiever te kunnen behandelen.
Invloed op het dagelijks leven
Leven met ALS verandert het dagelijkse leven ingrijpend door het geleidelijke verlies van spierfunctie en motorische controle. Eenvoudige handelingen zoals lopen, voorwerpen oprapen en zelfs spreken worden steeds moeilijker naarmate de ziekte voortschrijdt. Taken die ooit vanzelfsprekend waren, vereisen aanzienlijke inspanning of kunnen onmogelijk nog zelfstandig worden uitgevoerd. Omdat het vermogen om te bewegen en te communiceren afneemt, hebben ALS-patiënten vaak hulpmiddelen en aangepaste technieken nodig om een bepaald niveau van functionaliteit en betrokkenheid bij hun dagelijkse routines te behouden.
Ondersteuning voor patiënten en familie
Leven met ALS vraagt om een stevig ondersteuningsnetwerk voor zowel patiënten als hun familie. Patiënten hebben fysieke, emotionele en psychologische steun nodig om de uitdagingen van de ziekte aan te kunnen. Dit omvat hulp bij dagelijkse activiteiten, het beheren van medische behoeften en het omgaan met emotioneel welzijn.
Familieleden en verzorgers spelen een cruciale rol bij het bieden van deze ondersteuning terwijl ze op zoek zijn naar antwoorden op de vraag "Wat is amyotrofische laterale sclerose?". Ook zij hebben hulpbronnen nodig om met de emotionele druk en praktische eisen van het zorgen om te gaan. Steungroepen, adviesdiensten en educatieve bronnen kunnen waardevolle hulp bieden bij het omgaan met de complexe aspecten van ALS.
Verhalen en perspectieven van mensen met ALS
Persoonlijke verhalen en perspectieven van mensen die leven met ALS bieden unieke inzichten in de reis die deze progressieve ziekte met zich meebrengt. Deze verhalen benadrukken de kracht, moed en veerkracht die nodig zijn om zich aan te passen aan de veranderingen en beperkingen die ALS met zich meebrengt. Ze bieden een platform voor het delen van ervaringen, uitdagingen en omgangsstrategieën. Het delen van deze verhalen bevordert het gemeenschapsgevoel onder ALS-patiënten, geeft hoop en zorgt voor een gevoel van verbondenheid dat individuen en families kan helpen zich minder geïsoleerd te voelen in hun strijd.
Sluit je aan bij onze snelgroeiende community en revolutioneer wetenschappelijke communicatie!
Ontdek hoe de Mind the Graph platform geeft wetenschappers een uitgebreide toolkit voor effectieve communicatie. Door visuals, data en informatie naadloos te integreren, vereenvoudigt Mind the Graph complexe wetenschappelijke concepten en maakt ze toegankelijk voor zowel experts als het bredere publiek.



Abonneer u op onze nieuwsbrief
Exclusieve inhoud van hoge kwaliteit over effectieve visuele
communicatie in de wetenschap.