
Når man taler om neurologiske lidelser, er der få tilstande, der er så indviklede og ødelæggende som amyotrofisk lateral sklerose, også kaldet ALS. Denne gådefulde sygdom, der ofte er synonym med Lou Gehrigs sygdom til ære for den berømte baseballspiller, hvis karriere på tragisk vis blev påvirket af sygdommen, er en indviklet gåde, der fortsat fanger opmærksomheden hos medicinske forskere, klinikere og den brede offentlighed.
Selvom ALS måske ikke har samme udbredte anerkendelse som visse andre neurologiske lidelser, er dens indvirkning på både de direkte berørte og deres kære dyb, ofte ledsaget af hjerteskærende følelser. Denne artikel med titlen "Hvad er amyotrofisk lateral sklerose og dens virkninger?" dykker ned i kernen af denne tragiske tilstand.
Desværre har det nylige tab af Bryan Randall, en fotograf og mangeårig partner til Sandra Bullock, endnu en gang sat fokus på ALS i offentligheden. Bryans død som 57-årig understregede sygdommens ubarmhjertige karakter. Desværre findes der ingen kur mod ALS, og efter diagnosen har personer typisk en gennemsnitlig forventet levetid på 2 til 5 år.
Hvad er amyotrofisk lateral sklerose?
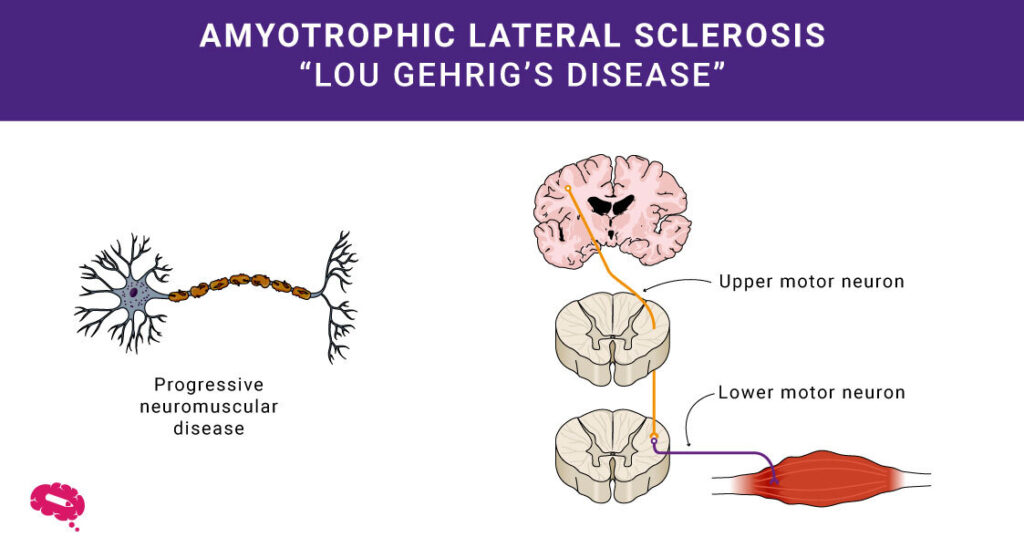
Lægevidenskaben afdækker ofte komplekse mysterier inden for menneskers sundhed. Et af dem er Amyotrofisk Lateral Sklerose, eller ALS. Du kender den måske som Lou Gehrigs sygdom. Denne sygdom påvirker hjernens og rygmarvens motorneuroner, som styrer menneskets muskler. Ved ALS nedbrydes disse neuroner, hvilket gør det svært at bevæge musklerne.
Definition af ALS
ALS, eller Amyotrofisk Lateral Sklerose, er en sygdom, der påvirker, hvordan menneskets muskler fungerer. Det gør, at musklerne med tiden bliver svagere, hvilket kan føre til problemer med at bevæge sig og udføre dagligdags opgaver.
Kort historie og baggrund
Historien om Amyotrofisk Lateral Sklerose (ALS) er en historie om opdagelser, der strækker sig over mere end et århundrede. Den begyndte i 1869, da den franske neurolog Jean-Martin Charcot først identificerede og beskrev sygdommen. ALS blev dog først kendt i 1939, da den legendariske baseballspiller Lou Gehrig blev diagnosticeret med den, hvilket førte til det mundrette navn "Lou Gehrigs sygdom".
Fremskridt i 1960'erne og 1970'erne belyste sygdommens kerne og afslørede den progressive degeneration af motorneuroner som dens underliggende mekanisme. I 1990'erne afslørede genetiske gennembrud forbindelser mellem ALS og gener som SOD1 og TDP-43, hvilket uddybede vores forståelse af dens genetiske komponenter.
Det 21. århundrede markerede vigtige milepæle, herunder den virale Ice Bucket Challenge, der skabte både opmærksomhed og ressourcer til ALS-forskning. I 2016 godkendte FDA det første ALS-specifikke lægemiddel, edaravone (Radicava), der tilbyder en ny tilgang til at bremse sygdommens udvikling.
Mens vi navigerer i 2020'erne, fortsætter den ubarmhjertige forskning med at udforske behandlingsmuligheder, de genetiske forviklinger ved ALS og de underliggende udløsende faktorer. Denne rejse understreger vores dedikation til at afdække kompleksiteten af ALS og lindre dens indvirkning på dem, der er berørt.
Vigtigheden af at forstå ALS
Forståelse af ALS spiller en vigtig rolle for forskellige individer og rækker ud over medicinske eksperter til at omfatte samfundet som helhed. Vigtigheden af denne forståelse stammer fra flere vigtige årsager:
For det første diskriminerer ALS ikke baseret på demografi, hvilket gør udbredt forståelse afgørende. Øget bevidsthed om sygdommen gør sundhedspersonale i stand til at identificere den i dens tidlige stadier, hvilket letter rettidig indgriben og pleje af de berørte personer.
For det andet er en dybere forståelse af ALS et kildevæld af håb for familier, der navigerer i dens udfordringer. Klar indsigt i sygdommens udvikling og tilgængelige behandlinger giver familierne mulighed for at planlægge fremad, tilpasse deres liv og træffe informerede valg.
Derudover får forskningsindsatsen momentum gennem forståelse. En solid forståelse af ALS' indviklede natur fremskynder søgningen efter potentielle terapier og interventioner. Med øget forståelse kan ressourcerne rettes mere effektivt mod effektive løsninger.
I sidste ende samler forståelsen af ALS en kollektiv front. Familier, venner, plejepersonale, forskere og det bredere samfund går sammen om at tackle denne udfordring. Gennem omfattende forståelse fremmes en stærk bevægelse mod ALS, der forestiller sig en fremtid, hvor ALS er synonymt med modstandsdygtighed, solidaritet og fremskridt.
Forståelse af sygdommen
Amyotrofisk lateral sklerose (ALS) er kendt for at være en kompleks sygdom. Her er nogle vigtige detaljer at kaste lys over:
Symptomer og tidlige tegn
For at opdage ALS tidligt er det vigtigt at identificere de karakteristiske symptomer. De første indikatorer involverer ofte svækkede muskler, især i lemmerne. Andre potentielle tegn omfatter ændringer i tale, muskeltrækninger og træthedsfornemmelser.
Årsager og risikofaktorer
Den præcise årsag til ALS er ikke helt klarlagt, men man mener, at det er et resultat af en kombination af genetiske og miljømæssige faktorer. Her er nogle af de kendte årsager og risikofaktorer, der er forbundet med ALS:
- Genetiske faktorer: En lille procentdel (ca. 5-10%) af ALS-tilfældene er arvelige, hvilket betyder, at de skyldes mutationer i specifikke gener. Det mest velkendte gen, der er forbundet med familiær ALS, er C9orf72-genmutationen. Mutationer i andre gener som SOD1, TARDBP og FUS er også blevet sat i forbindelse med ALS. Disse genetiske mutationer kan føre til ophobning af unormale proteiner i neuronerne, hvilket forårsager skader og celledød.
- Miljømæssige faktorer: Mens genetiske faktorer spiller en væsentlig rolle i familiære tilfælde af ALS, menes miljømæssige faktorer også at bidrage til udviklingen af sygdommen. Eksponering for visse toksiner og kemikalier, såsom tungmetaller, pesticider og industrielle forurenende stoffer, er blevet foreslået som potentielle risikofaktorer.
- Alder: ALS udvikler sig typisk hos voksne mellem 40 og 70 år, og gennemsnitsalderen for sygdomsdebut er omkring 55-60 år. Det kan dog forekomme i alle aldre.
- Køn: ALS er lidt mere almindelig hos mænd end hos kvinder. Årsagerne til denne kønsforskel er ikke helt klarlagt.
- Race og etnicitet: ALS forekommer i alle racer og etniske grupper, men det ser ud til at være mere almindeligt blandt kaukasiere sammenlignet med andre racegrupper.
- Rygning: Der er noget, der tyder på, at rygning kan øge risikoen for at udvikle ALS. Rygning er blevet forbundet med oxidativ stress og inflammation, som menes at spille en rolle i sygdommens udvikling.
- Fysisk aktivitet: Der er noget, der tyder på, at personer, der tidligere har været meget fysisk aktive, kan have en let øget risiko for ALS. Forholdet mellem fysisk aktivitet og ALS-risiko er dog ikke fuldt forstået og kræver yderligere forskning.
- Hovedtraume: Der har været antydninger af, at en historie med traumatiske hovedskader kan være forbundet med en let øget risiko for ALS. Men sammenhængen mellem hovedtraumer og ALS er stadig under undersøgelse.
Hvordan ALS påvirker kroppen
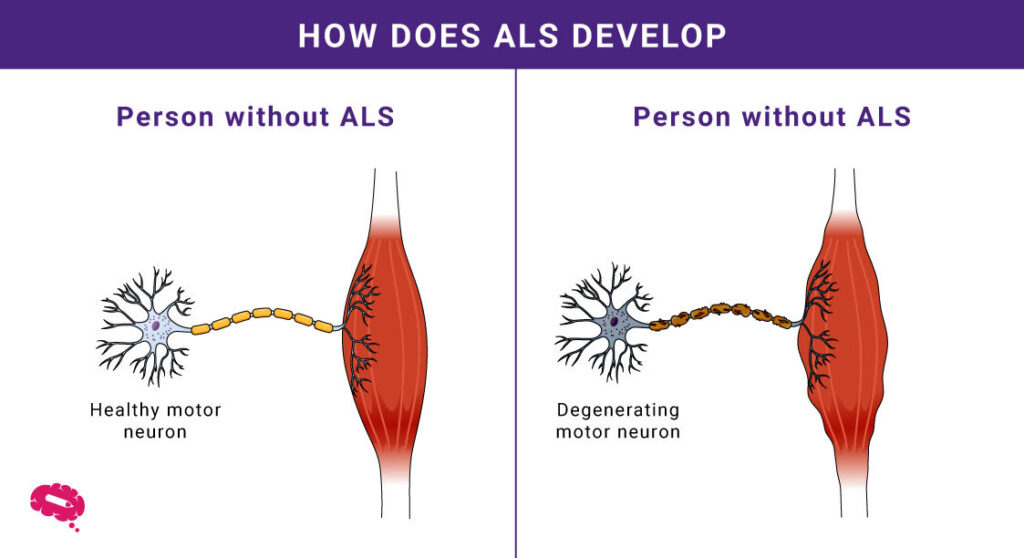
- Degeneration af motorneuroner: ALS rammer motorneuroner og forårsager muskelsvaghed og tab af kontrol.
- Muskelsvaghed: Gradvis degeneration fører til vanskeligheder med at bevæge sig og udføre dagligdags opgaver.
- Tale- og synkeproblemer: Tale- og synkemusklerne påvirkes, så man taler utydeligt og har svært ved at spise.
- Vejrtrækningsproblemer: Åndedrætsmusklerne svækkes, hvilket fører til åndedrætsbesvær.
- Progressiv lammelse: Med tiden påvirker den tiltagende lammelse mobilitet og uafhængighed.
- Kognitive forandringer: Nogle oplever ændringer i hukommelse og beslutningstagning, især ved ALS med frontotemporal demens.
- Sansefunktion intakt: Følelser som berøring og syn forbliver normalt upåvirkede.
- Livstruende komplikationer: Åndedrætssvigt i fremskredne stadier kan være livstruende.
Diagnose og testning
ALS-diagnosen er en omfattende proces, der omfatter klinisk evaluering, vurdering af sygehistorien, neurologisk undersøgelse og udelukkelse af alternative tilstande. Den er afhængig af en række undersøgelser, herunder elektromyografi (EMG), nerveledningsundersøgelser (NCS) og billeddannelse, som tilsammen evaluerer nerve- og muskelfunktion. Det er bemærkelsesværdigt, at ALS ikke har en eneste definitiv test, men at diagnosen snarere afhænger af en sammenlægning af kliniske observationer og resultater fra forskellige tests.
Diagnostiske procedurer
- Gennemgang af sygehistorien for at forstå symptomerne og deres udvikling.
- Foretage en neurologisk vurdering for at evaluere muskelstyrke, reflekser og koordination.
- Elektromyografi (EMG) måler muskelaktivitet og opdager nerveproblemer.
- Nerveledningsundersøgelser (NCS) vurderer nervefunktion og respons.
Udfordringer ved diagnosticering
- ALS-symptomer overlapper med andre sygdomme, hvilket fører til fejldiagnosticering eller forsinket diagnose.
- Manglen på specifikke biomarkører kan forhindre en tidlig og præcis diagnose.
- Progression og symptomvariation gør diagnosen udfordrende.
At skelne ALS fra andre sygdomme
I modsætning til multipel sklerose (MS), som primært påvirker centralnervesystemets myelinskede, er ALS rettet mod motorneuroner, der er ansvarlige for frivillig muskelkontrol.
Mens primær lateral sklerose (PLS) deler visse karakteristika med ALS, involverer den overvejende øvre motorneuroner, til forskel fra ALS's kombinerede øvre og nedre motorneuron involvering.
Derudover er det afgørende at skelne mellem ALS og spinal muskelatrofi (SMA) på grund af deres fælles påvirkning af motorneuroner; SMA påvirker dog primært lavere motorneuroner i modsætning til ALS. Evnen til at skelne ALS fra disse sygdomme er afgørende for en præcis diagnose og skræddersyede behandlingsstrategier.
Behandling og håndtering
Behandling og håndtering af ALS omfatter en multifacetteret tilgang. De tilgængelige behandlingsmuligheder sigter mod at lindre symptomer, bremse sygdommens udvikling og forbedre livskvaliteten for personer med ALS.
Tilgængelige behandlingsmuligheder
- Hovedfokus for ALS-behandling er at afhjælpe symptomer, forbedre livskvaliteten og bremse sygdommens udvikling.
- Medicin som riluzol og edaravone er godkendt til potentielt at forlænge overlevelsen og forsinke progressionen.
- Baclofen og andre lægemidler kan hjælpe med at håndtere muskelspasticitet, mens medicin som antikolinergika afhjælper overdreven spytproduktion.
- Hjælpemidler, såsom ikke-invasive respiratorer og ernæringssonder, hjælper med vejrtrækning og ernæring, når muskelfunktionen aftager.
Håndtering af symptomer og livskvalitet
- Fysio- og ergoterapi hjælper med at bevare muskelstyrke, mobilitet og uafhængighed.
- Logopædi hjælper med kommunikation og synkebesvær.
- Støttende terapier, som ernæringsrådgivning, psykologisk rådgivning og støttegrupper, hjælper med at håndtere følelsesmæssige og praktiske udfordringer.
- Palliativ pleje og hospice forbedrer komforten og livskvaliteten i fremskredne stadier.
Igangværende forskning og fremtidige behandlingsformer
- Forskere fortsætter med at undersøge ALS' underliggende årsager og potentielle behandlingsmål.
- Genterapi har til formål at korrigere genetiske mutationer, der er ansvarlige for familiær ALS.
- Stamcelleforskning undersøger brugen af stamceller til at erstatte beskadigede motorneuroner.
- Innovative tilgange omfatter målretning mod unormal proteinakkumulering og inflammation i ALS-ramte neuroner.
- Udvikling af biomarkører kan hjælpe med at stille en tidlig diagnose og følge sygdommens udvikling.
- Man søger fortsat efter nye terapeutiske midler og strategier, der kan give mere effektive behandlinger af ALS.
Indvirkning på det daglige liv
At leve med ALS er en stor omvæltning i dagligdagen på grund af det gradvise tab af muskelfunktion og motorisk kontrol. Simple handlinger som at gå, samle genstande op og endda tale bliver stadig sværere, efterhånden som sygdommen skrider frem. Opgaver, der engang blev taget for givet, kræver en betydelig indsats eller kan blive umulige at udføre selvstændigt. Efterhånden som evnen til at bevæge sig og kommunikere aftager, har personer med ALS ofte brug for hjælpemidler og tilpasningsteknikker for at opretholde et vist niveau af funktionalitet og engagement i deres daglige rutiner.
Støtte til patienter og familier
At leve med ALS kræver et robust støttenetværk for både patienter og deres familier. Patienter har brug for fysisk, følelsesmæssig og psykologisk støtte til at navigere i sygdommens udfordringer. Det omfatter hjælp til daglige aktiviteter, håndtering af medicinske behov og håndtering af følelsesmæssigt velbefindende.
Familier og omsorgspersoner spiller en afgørende rolle i at yde denne støtte, mens de søger svar på spørgsmålet "Hvad er amyotrofisk lateral sklerose?" De har også brug for ressourcer til at klare den følelsesmæssige belastning og de praktiske krav, der er forbundet med at være pårørende. Støttegrupper, rådgivningstjenester og uddannelsesmæssige ressourcer kan give værdifuld hjælp til at håndtere kompleksiteten ved ALS.
Historier og perspektiver fra personer med ALS
Personlige historier og perspektiver fra personer, der lever med ALS, giver et unikt indblik i den rejse, det er at stå over for denne progressive sygdom. Disse historier fremhæver den styrke, det mod og den modstandsdygtighed, der kræves for at tilpasse sig de forandringer og begrænsninger, som ALS medfører. De giver en platform til at dele erfaringer, udfordringer og mestringsstrategier. At dele disse fortællinger fremmer en følelse af fællesskab blandt dem, der er berørt af ALS, inspirerer til håb og giver en følelse af samhørighed, der kan hjælpe enkeltpersoner og familier med at føle sig mindre isolerede i deres kamp.
Deltag i vores hurtigt voksende fællesskab for at revolutionere videnskabelig kommunikation!
Opdag, hvordan Mind the Graph platformen giver forskere en omfattende værktøjskasse til effektiv kommunikation. Ved problemfrit at integrere grafik, data og information forenkler Mind the Graph komplekse videnskabelige koncepter og gør dem tilgængelige for både eksperter og det brede publikum.



Tilmeld dig vores nyhedsbrev
Eksklusivt indhold af høj kvalitet om effektiv visuel
kommunikation inden for videnskab.