
När man diskuterar neurologiska sjukdomar är det få tillstånd som är så komplicerade och förödande som amyotrofisk lateralskleros, mer allmänt kallad ALS. Denna gåtfulla sjukdom, ofta synonym med Lou Gehrigs sjukdom efter den berömde basebollspelaren vars karriär tragiskt påverkades av sjukdomen, är en invecklad gåta som fortsätter att fängsla medicinska forskare, läkare och den bredare allmänheten.
Även om ALS kanske inte är lika känt som vissa andra neurologiska sjukdomar, har sjukdomen en djupgående inverkan på både de direkt drabbade och deras anhöriga, och åtföljs ofta av hjärtskärande känslor. Denna artikel, med titeln "Vad är amyotrofisk lateralskleros och dess effekter?", fördjupar sig i kärnan av detta tragiska tillstånd.
Tyvärr har den senaste förlusten av Bryan Randall, fotograf och mångårig partner till Sandra Bullock, återigen satt fokus på ALS i allmänhetens ögon. Bryans bortgång vid 57 års ålder understryker sjukdomens oförlåtande karaktär. Tyvärr finns det inget botemedel mot ALS, och efter diagnos har individer i genomsnitt en förväntad livslängd på 2 till 5 år.
Vad är amyotrofisk lateral skleros?
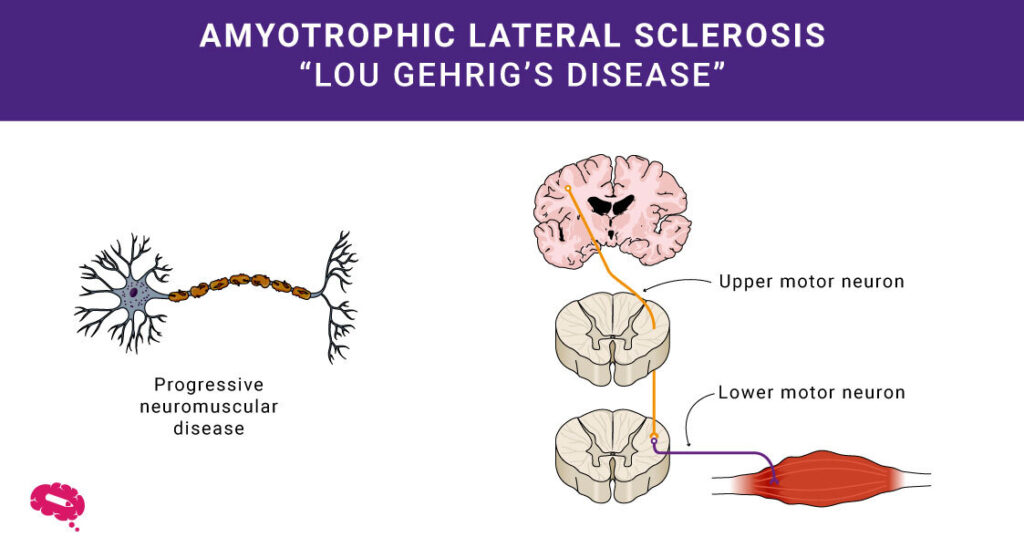
Medicinsk vetenskap avslöjar ofta komplexa mysterier inom området för människors hälsa. Ett av dem är amyotrofisk lateralskleros, eller ALS. Du kanske känner till det som Lou Gehrigs sjukdom. Denna sjukdom påverkar hjärnans och ryggmärgens motorneuron, som styr människans muskler. Vid ALS bryts dessa nervceller ned, vilket gör det svårt att röra musklerna.
Definition av ALS
ALS, eller amyotrofisk lateralskleros, är en sjukdom som påverkar hur de mänskliga musklerna fungerar. Detta gör att de mänskliga musklerna blir svagare med tiden, vilket kan leda till problem med att röra sig och utföra vardagliga uppgifter.
Kort historik och bakgrund
Amyotrofisk lateralskleros (ALS) är en upptäcktshistoria som sträcker sig över mer än ett sekel. Den började 1869 när den franske neurologen Jean-Martin Charcot först identifierade och beskrev sjukdomen. ALS blev dock mer känt 1939 när den legendariske basebollspelaren Lou Gehrig diagnostiserades med sjukdomen, vilket ledde till det vardagliga namnet "Lou Gehrigs sjukdom".
Framsteg under 1960- och 1970-talen belyste sjukdomens kärna och avslöjade den progressiva degenerationen av motorneuroner som dess underliggande mekanism. På 1990-talet avslöjade genetiska genombrott kopplingar mellan ALS och gener som SOD1 och TDP-43, vilket fördjupade vår förståelse av dess genetiska komponenter.
2000-talet har inneburit viktiga milstolpar, bland annat den virala Ice Bucket Challenge som ökade både medvetenheten och resurserna för ALS-forskning. År 2016 godkände FDA det första ALS-specifika läkemedlet, edaravone (Radicava), som erbjuder en ny metod för att bromsa sjukdomens utveckling.
Under 2020-talet fortsätter den obevekliga forskningen med att utforska behandlingsmöjligheter, de genetiska svårigheterna med ALS och de underliggande utlösande faktorerna. Denna resa understryker vårt engagemang för att ta reda på komplexiteten i ALS och lindra dess inverkan på de drabbade.
Vikten av att förstå ALS
Att förstå ALS spelar en viktig roll för olika individer, och sträcker sig bortom medicinska experter till att omfatta samhället som helhet. Det finns flera viktiga skäl till att det är så viktigt att förstå sjukdomen:
För det första diskriminerar ALS inte baserat på demografiska faktorer, vilket gör det viktigt med en bred förståelse. Ökad medvetenhet om sjukdomen gör det möjligt för sjukvårdspersonal att identifiera den i ett tidigt skede, vilket underlättar tidiga insatser och vård för drabbade personer.
För det andra innebär en djupare förståelse av ALS en källa till hopp för familjer som står inför stora utmaningar. Tydliga insikter om sjukdomens förlopp och tillgängliga behandlingar ger familjerna möjlighet att planera framåt, anpassa sina liv och göra välgrundade val.
Dessutom får forskningsinsatserna fart genom förståelse. En gedigen förståelse av ALS komplicerade natur påskyndar sökandet efter potentiella terapier och interventioner. Med ökad förståelse kan resurserna riktas mer effektivt mot effektiva lösningar.
I slutändan samlar förståelsen av ALS en kollektiv front. Familjer, vänner, vårdgivare, forskare och samhället i stort förenas för att ta sig an denna utmaning. Genom en heltäckande förståelse främjas en kraftfull rörelse mot ALS, och vi ser framför oss en framtid där ALS är synonymt med motståndskraft, solidaritet och framsteg.
Att förstå sjukdomen
Amyotrofisk lateralskleros (ALS) är välkänd för att vara en komplex sjukdom. Här är några viktiga detaljer att belysa:
Symtom och tidiga tecken
För att upptäcka ALS tidigt är det viktigt att identifiera de karakteristiska symtomen. De första tecknen är ofta försvagade muskler, särskilt i armar och ben. Andra potentiella tecken är förändringar i talet, muskelryckningar och trötthetskänslor.
Orsaker och riskfaktorer
Den exakta orsaken till ALS är inte helt klarlagd, men man tror att den beror på en kombination av genetiska faktorer och miljöfaktorer. Här är några av de kända orsaker och riskfaktorer som förknippas med ALS:
- Genetiska faktorer: En liten andel (cirka 5-10%) av ALS-fallen är ärftliga, vilket innebär att de orsakas av mutationer i specifika gener. Den mest välkända genen som förknippas med familjär ALS är C9orf72-genmutationen. Mutationer i andra gener som SOD1, TARDBP och FUS har också kopplats till ALS. Dessa genetiska mutationer kan leda till ackumulering av onormala proteiner i nervcellerna, vilket orsakar skador och celldöd.
- Miljöfaktorer: Även om genetiska faktorer spelar en viktig roll i familjära fall av ALS, anses miljöfaktorer också kunna bidra till utvecklingen av sjukdomen. Exponering för vissa toxiner och kemikalier, t.ex. tungmetaller, bekämpningsmedel och industriföroreningar, har föreslagits som potentiella riskfaktorer.
- Ålder: ALS utvecklas vanligtvis hos vuxna mellan 40 och 70 års ålder, med en genomsnittlig debutålder på cirka 55-60 år. Sjukdomen kan dock uppträda i alla åldrar.
- Kön: ALS är något vanligare hos män än hos kvinnor. Orsakerna till denna skillnad mellan könen är inte helt klarlagda.
- Ras och etnicitet: ALS förekommer i alla ras- och etniska grupper, men verkar vara vanligare bland kaukasier jämfört med andra ras- och etniska grupper.
- Rökning: Det finns belägg för att rökning kan öka risken för att utveckla ALS. Rökning har förknippats med oxidativ stress och inflammation, som anses spela en roll i sjukdomsförloppet.
- Fysisk aktivitet: Det finns vissa belägg för att personer som tidigare har varit mycket fysiskt aktiva kan ha en något ökad risk för ALS. Sambandet mellan fysisk aktivitet och risken för ALS är dock inte helt klarlagt och kräver ytterligare forskning.
- Skalltrauma: Det har antytts att en historia av traumatiska huvudskador kan vara förknippad med en något ökad risk för ALS. Sambandet mellan huvudtrauma och ALS håller dock fortfarande på att undersökas.
Hur ALS påverkar kroppen
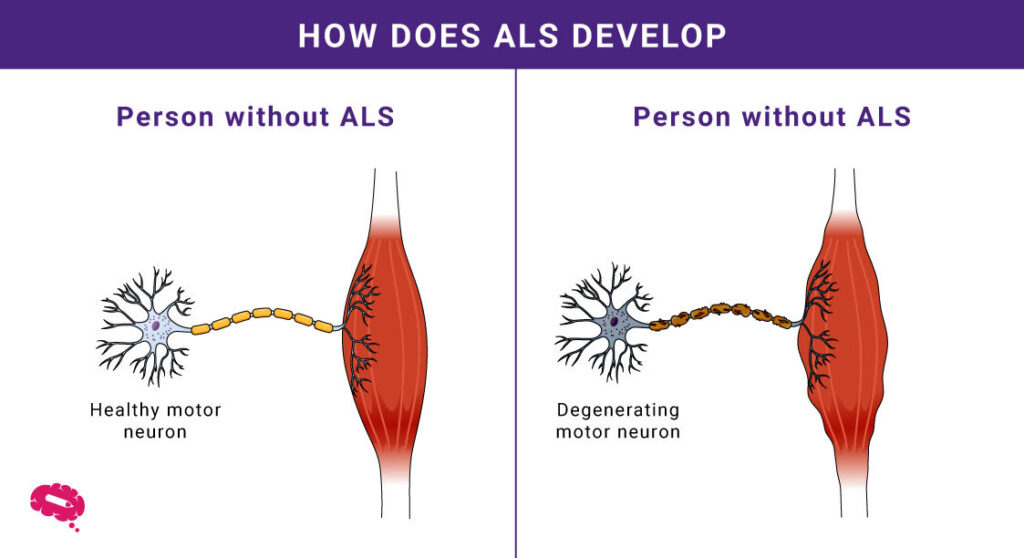
- Degeneration av motorneuroner: ALS drabbar motorneuroner och orsakar muskelsvaghet och förlust av kontroll.
- Muskelsvaghet: Gradvis degeneration leder till svårigheter att röra sig och utföra vardagliga sysslor.
- Tal- och sväljproblem: Musklerna för tal och sväljning påverkas, vilket orsakar sluddrande och svårigheter att äta.
- Problem med andningen: Andningsmusklerna försvagas, vilket leder till andningssvårigheter.
- Progressiv förlamning: Med tiden påverkar en tilltagande förlamning rörelseförmågan och oberoendet.
- Kognitiva förändringar: Vissa upplever förändringar i minne och beslutsfattande, särskilt vid ALS med frontotemporal demens.
- Sensorisk funktion intakt: Sinnesförnimmelser som känsel och syn påverkas vanligtvis inte.
- Livshotande komplikationer: Andningssvikt i avancerade stadier kan vara livshotande.
Diagnos och testning
ALS-diagnosen är en omfattande process som omfattar klinisk utvärdering, bedömning av sjukdomshistoria, neurologisk undersökning och uteslutning av alternativa tillstånd. Den bygger på en rad olika undersökningar, inklusive elektromyografi (EMG), nervledningsstudier (NCS) och bilddiagnostik, som tillsammans utvärderar nerv- och muskelfunktionen. Det är anmärkningsvärt att ALS saknar ett enda definitivt test, utan diagnosen beror snarare på en sammanslagning av kliniska observationer och resultat från olika tester.
Diagnostiska förfaranden
- Genomgång av sjukdomshistoria för att förstå symtom och deras utveckling.
- Genomföra en neurologisk bedömning för att utvärdera muskelstyrka, reflexer och koordination.
- Elektromyografi (EMG) mäter muskelaktivitet och upptäcker nervproblem.
- Nervledningsstudier (NCS) utvärderar nervfunktion och nervrespons.
Utmaningar vid diagnostisering
- ALS symtom överlappar med andra sjukdomar, vilket leder till feldiagnos eller fördröjd diagnos.
- Bristen på specifika biomarkörer kan hindra tidig och korrekt diagnos.
- Progression och symtomvariation gör det svårt att ställa diagnos.
Att skilja ALS från andra sjukdomar
Till skillnad från multipel skleros (MS), som främst påverkar myelinskidan i det centrala nervsystemet, riktar sig ALS mot motorneuroner som ansvarar för viljestyrd muskelkontroll.
Primär lateral skleros (PLS) har vissa likheter med ALS, men drabbar främst de övre motorneuronerna, till skillnad från ALS där både de övre och nedre motorneuronerna är drabbade.
Dessutom är det viktigt att skilja ALS från spinal muskelatrofi (SMA) eftersom de båda sjukdomarna påverkar motorneuronerna. SMA påverkar dock främst de lägre motorneuronerna, till skillnad från ALS. Förmågan att skilja ALS från dessa sjukdomar är avgörande för en korrekt diagnos och skräddarsydda behandlingsstrategier.
Behandling och hantering
Behandling och hantering av ALS omfattar ett mångfacetterat tillvägagångssätt. Tillgängliga behandlingsalternativ syftar till att lindra symtomen, bromsa sjukdomsförloppet och förbättra livskvaliteten för personer med ALS.
Tillgängliga behandlingsalternativ
- Huvudsyftet med ALS-behandling är att lindra symtomen, förbättra livskvaliteten och bromsa sjukdomsförloppet.
- Läkemedel som riluzole och edaravone är godkända för att potentiellt förlänga överlevnaden och fördröja sjukdomsprogressionen.
- Baklofen och andra läkemedel kan hjälpa till att hantera muskelspasticitet, medan läkemedel som antikolinergika behandlar överdriven salivproduktion.
- Hjälpmedel, som icke-invasiva ventilatorer och sondmatning, underlättar andning och nutrition när muskelfunktionen försämras.
Symtomhantering och livskvalitet
- Fysioterapi och arbetsterapi hjälper till att bibehålla muskelstyrka, rörlighet och självständighet.
- Logopedi hjälper till med kommunikation och sväljsvårigheter.
- Stödjande behandlingar, som kostrådgivning, psykologisk rådgivning och stödgrupper, hjälper till att hantera känslomässiga och praktiska utmaningar.
- Palliativ vård och hospice förbättrar komforten och livskvaliteten i avancerade stadier.
Pågående forskning och framtida behandlingar
- Forskare fortsätter att undersöka de bakomliggande orsakerna till ALS och potentiella behandlingsmål.
- Syftet med genterapi är att korrigera genetiska mutationer som orsakar familjär ALS.
- Stamcellsforskning undersöker möjligheten att använda stamceller för att ersätta skadade motorneuron.
- Innovativa metoder inkluderar inriktning på onormal proteinackumulering och inflammation i nervceller som drabbats av ALS.
- Att utveckla biomarkörer skulle kunna bidra till tidig diagnos och till att följa sjukdomsutvecklingen.
- Sökandet efter nya terapeutiska medel och strategier fortsätter för att få fram mer effektiva behandlingar av ALS.
Påverkan på det dagliga livet
Att leva med ALS innebär stora förändringar i det dagliga livet på grund av den gradvisa förlusten av muskelfunktion och motorisk kontroll. Enkla handlingar som att gå, plocka upp föremål och till och med att tala blir allt svårare ju längre sjukdomen fortskrider. Uppgifter som en gång togs för givna kräver stor ansträngning eller kan bli omöjliga att utföra på egen hand. När förmågan att röra sig och kommunicera försämras behöver personer med ALS ofta hjälpmedel och anpassningstekniker för att upprätthålla en viss nivå av funktionalitet och engagemang i sina dagliga rutiner.
Stöd till patienter och anhöriga
Att leva med ALS kräver ett robust stödnätverk för både patienter och deras familjer. Patienterna behöver fysiskt, känslomässigt och psykologiskt stöd för att klara av de utmaningar som sjukdomen innebär. Det kan handla om hjälp med dagliga aktiviteter, att hantera medicinska behov och att ta hand om det känslomässiga välbefinnandet.
Familjer och vårdgivare spelar en avgörande roll när det gäller att ge detta stöd samtidigt som de söker svar på frågan "Vad är amyotrofisk lateralskleros?" De behöver också resurser för att klara av den känslomässiga påfrestning och de praktiska krav som vårdandet innebär. Stödgrupper, rådgivningstjänster och utbildningsresurser kan ge värdefull hjälp med att hantera den komplexa ALS-sjukdomen.
Berättelser och perspektiv från personer med ALS
Personliga berättelser och perspektiv från personer som lever med ALS ger unika insikter i hur det är att möta denna progressiva sjukdom. Dessa berättelser belyser den styrka, det mod och den uthållighet som krävs för att anpassa sig till de förändringar och begränsningar som ALS medför. De utgör en plattform för att dela erfarenheter, utmaningar och strategier. Att dela dessa berättelser främjar en känsla av gemenskap bland dem som drabbats av ALS, inspirerar till hopp och ger en känsla av samhörighet som kan hjälpa individer och familjer att känna sig mindre isolerade i sin kamp.
Gå med i vår snabbväxande gemenskap för att revolutionera vetenskaplig kommunikation!
Upptäck hur Mind the Graph plattformen ger forskare en omfattande verktygslåda för effektiv kommunikation. Genom att sömlöst integrera grafik, data och information förenklar Mind the Graph komplexa vetenskapliga koncept och gör dem tillgängliga för både experter och en bredare publik.



Prenumerera på vårt nyhetsbrev
Exklusivt innehåll av hög kvalitet om effektiv visuell
kommunikation inom vetenskap.