
Når man snakker om nevrologiske lidelser, er det få tilstander som er så kompliserte og ødeleggende som amyotrofisk lateral sklerose, ofte omtalt som ALS. Denne gåtefulle sykdommen, som ofte blir synonymt med Lou Gehrigs sykdom til ære for den berømte baseballspilleren hvis karriere ble tragisk påvirket av den, er en intrikat gåte som fortsetter å fange oppmerksomheten til medisinske forskere, klinikere og det brede publikum.
Selv om ALS kanskje ikke er like kjent som visse andre nevrologiske lidelser, har sykdommen en dyp innvirkning på både de som rammes direkte og deres nærmeste, og er ofte ledsaget av hjerteskjærende følelser. Denne artikkelen, med tittelen "Hva er amyotrofisk lateral sklerose og dens konsekvenser?", tar for seg kjernen i denne tragiske tilstanden.
Dessverre har den nylige bortgangen til Bryan Randall, fotograf og mangeårig partner av Sandra Bullock, nok en gang satt fokus på ALS i offentligheten. Bryans bortgang i en alder av 57 år understreket sykdommens nådeløse karakter. Dessverre finnes det ingen kur mot ALS, og etter at diagnosen er stilt, har den syke vanligvis en gjennomsnittlig forventet levetid på to til fem år.
Hva er amyotrofisk lateral sklerose?
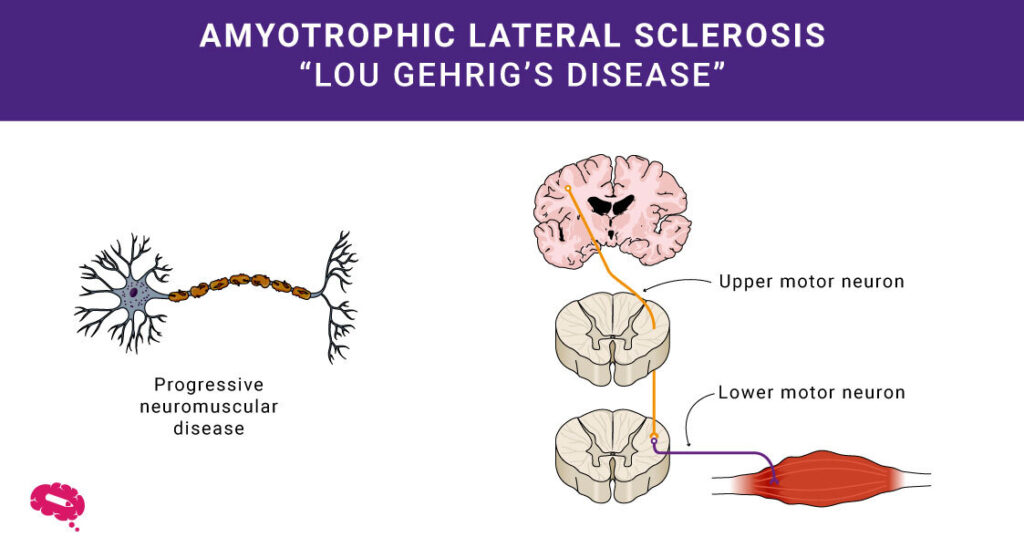
Den medisinske vitenskapen avdekker ofte komplekse mysterier når det gjelder menneskers helse. Et av dem er amyotrofisk lateral sklerose, eller ALS. Du kjenner den kanskje som Lou Gehrigs sykdom. Sykdommen rammer hjernens og ryggmargens motoriske nevroner, som kontrollerer musklene. Ved ALS brytes disse nevronene ned, noe som gjør det vanskelig å bevege musklene.
Definisjon av ALS
ALS, eller amyotrofisk lateral sklerose, er en sykdom som påvirker hvordan musklene fungerer. Dette gjør at musklene blir svakere over tid, noe som kan føre til problemer med å bevege seg og utføre dagligdagse oppgaver.
Kort historikk og bakgrunn
Historien om amyotrofisk lateral sklerose (ALS) er en oppdagelseshistorie som strekker seg over mer enn hundre år. Den begynte i 1869, da den franske nevrologen Jean-Martin Charcot først identifiserte og beskrev sykdommen. ALS ble imidlertid mer kjent i 1939, da den legendariske baseballspilleren Lou Gehrig fikk diagnosen, noe som førte til det folkelige navnet "Lou Gehrigs sykdom".
Fremskritt på 1960- og 1970-tallet belyste sykdommens kjerne, og avslørte den progressive degenerasjonen av motoriske nevroner som den underliggende mekanismen. På 1990-tallet avdekket genetiske gjennombrudd koblinger mellom ALS og gener som SOD1 og TDP-43, noe som ga oss en dypere forståelse av de genetiske komponentene.
Det 21. århundret har vært preget av viktige milepæler, blant annet den virale Ice Bucket Challenge som økte både bevisstheten om og ressursene til ALS-forskningen. I 2016 godkjente FDA det første ALS-spesifikke legemiddelet, edaravone (Radicava), som tilbyr en ny metode for å bremse sykdomsutviklingen.
På vei inn i 2020-tallet fortsetter forskningen med å utforske behandlingsmuligheter, de genetiske detaljene ved ALS og de underliggende utløsende faktorene. Denne reisen understreker vårt engasjement for å avdekke kompleksiteten i ALS og lindre konsekvensene av sykdommen for dem som er rammet.
Viktigheten av å forstå ALS
Forståelsen av ALS er av stor betydning for mange mennesker, ikke bare for medisinske eksperter, men også for samfunnet som helhet. Det er flere viktige grunner til at denne forståelsen er så viktig:
For det første diskriminerer ikke ALS på bakgrunn av demografiske forhold, noe som gjør det viktig å øke kunnskapen om sykdommen. Økt kunnskap om sykdommen gjør helsepersonell i stand til å identifisere den på et tidlig stadium, noe som gjør det lettere å gripe inn i tide og gi behandling til de som er rammet.
For det andre gir en dypere forståelse av ALS håp for familier som står midt oppe i utfordringene. Tydelig innsikt i sykdommens progresjon og tilgjengelige behandlinger gir familiene mulighet til å planlegge fremover, tilpasse livene sine og ta informerte valg.
I tillegg får forskningsinnsatsen fart gjennom forståelse. En solid forståelse av ALS' intrikate natur fremskynder jakten på potensielle behandlingsformer og intervensjoner. Med økt forståelse kan ressursene rettes mer effektivt mot effektive løsninger.
Til syvende og sist samler forståelsen av ALS en kollektiv front. Familier, venner, omsorgspersoner, forskere og samfunnet for øvrig går sammen om å håndtere denne utfordringen. Gjennom en helhetlig forståelse skapes det en kraftfull bevegelse mot ALS, og vi ser for oss en fremtid der ALS er synonymt med motstandsdyktighet, solidaritet og fremgang.
Forståelse av sykdommen
Amyotrofisk lateral sklerose (ALS) er kjent for å være en kompleks sykdom. Her er noen viktige detaljer for å kaste lys over den:
Symptomer og tidlige tegn
For å oppdage ALS tidlig er det viktig å identifisere de karakteristiske symptomene. De første tegnene er ofte svekket muskulatur, spesielt i ekstremitetene. Andre mulige tegn kan være taleforstyrrelser, muskelrykninger og tretthetsfølelse.
Årsaker og risikofaktorer
Den eksakte årsaken til ALS er ikke helt klarlagt, men det antas å være et resultat av en kombinasjon av genetiske og miljømessige faktorer. Her er noen av de kjente årsakene og risikofaktorene som er forbundet med ALS:
- Genetiske faktorer: En liten andel (ca. 5-10%) av ALS-tilfellene er arvelige, noe som betyr at de skyldes mutasjoner i spesifikke gener. Det mest kjente genet som er forbundet med familiær ALS, er mutasjonen i C9orf72-genet. Mutasjoner i andre gener som SOD1, TARDBP og FUS har også blitt knyttet til ALS. Disse genmutasjonene kan føre til at unormale proteiner hoper seg opp i nevronene og forårsaker skader og celledød.
- Miljøfaktorer: Selv om genetiske faktorer spiller en viktig rolle i familiære tilfeller av ALS, antas også miljøfaktorer å bidra til sykdomsutviklingen. Eksponering for visse giftstoffer og kjemikalier, som tungmetaller, plantevernmidler og industriforurensning, har blitt foreslått som potensielle risikofaktorer.
- Alder: ALS utvikles vanligvis hos voksne mellom 40 og 70 år, med en gjennomsnittlig debutalder på rundt 55-60 år. Sykdommen kan imidlertid oppstå i alle aldre.
- Kjønn: ALS er noe vanligere hos menn enn hos kvinner. Årsakene til denne kjønnsforskjellen er ikke helt klarlagt.
- Rase og etnisitet: ALS forekommer i alle rase- og etniske grupper, men ser ut til å være vanligere blant kaukasiere enn i andre rasegrupper.
- Røyking: Mye tyder på at røyking kan øke risikoen for å utvikle ALS. Røyking har blitt assosiert med oksidativt stress og inflammasjon, som antas å spille en rolle i sykdomsutviklingen.
- Fysisk aktivitet: Noe tyder på at personer som tidligere har vært svært fysisk aktive, kan ha en noe økt risiko for ALS. Sammenhengen mellom fysisk aktivitet og ALS-risiko er imidlertid ikke helt klarlagt og krever ytterligere forskning.
- Hodetraumer: Det har vært antydet at en historie med traumatiske hodeskader kan være forbundet med en noe økt risiko for ALS. Sammenhengen mellom hodetraumer og ALS er imidlertid fortsatt under utforskning.
Hvordan ALS påvirker kroppen
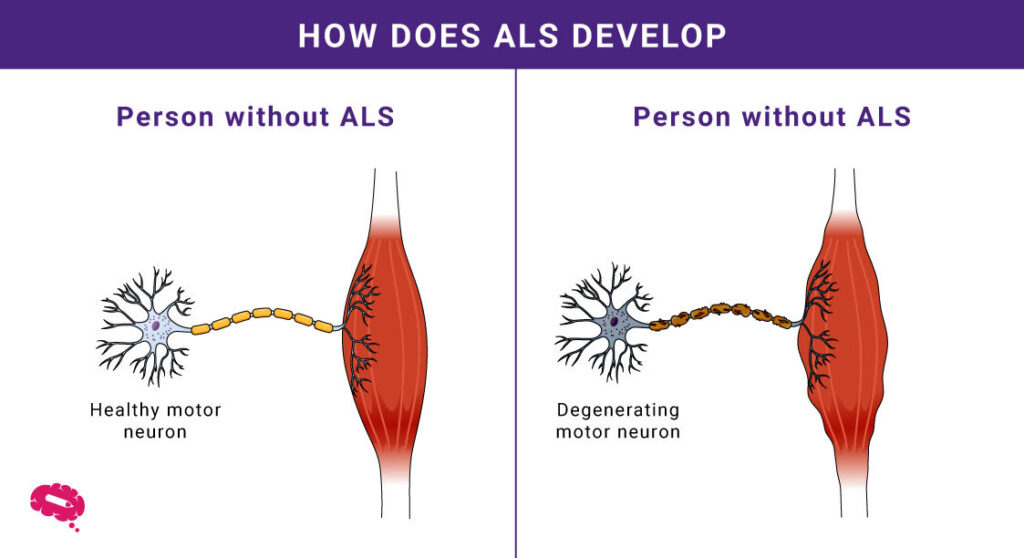
- Degenerasjon av motoriske nevroner: ALS angriper motoriske nevroner og forårsaker muskelsvakhet og tap av kontroll.
- Muskelsvakhet: Gradvis degenerasjon fører til vanskeligheter med å bevege seg og utføre dagligdagse oppgaver.
- Tale- og svelgeproblemer: Musklene som brukes til å snakke og svelge påvirkes, noe som fører til at man snakker utydelig og får problemer med å spise.
- Pusteproblemer: Åndedrettsmuskulaturen svekkes, noe som fører til pustevansker.
- Progressiv lammelse: Over tid vil økende lammelser påvirke bevegelighet og selvstendighet.
- Kognitive endringer: Noen opplever endringer i hukommelse og beslutningstaking, spesielt ved ALS med frontotemporal demens.
- Intakt sensorisk funksjon: Følelser som berøring og syn forblir vanligvis upåvirket.
- Livstruende komplikasjoner: Åndedrettssvikt i avanserte stadier kan være livstruende.
Diagnostisering og testing
ALS-diagnosen er en omfattende prosess som omfatter klinisk evaluering, anamnese, nevrologisk undersøkelse og utelukkelse av alternative tilstander. Diagnosen er avhengig av en rekke undersøkelser, inkludert elektromyografi (EMG), nerveledningsstudier (NCS) og bildediagnostikk, som til sammen evaluerer nerve- og muskelfunksjonen. Det er viktig å merke seg at det ikke finnes en eneste definitiv test for ALS, men at diagnosen er avhengig av en kombinasjon av kliniske observasjoner og resultater fra ulike tester.
Diagnostiske prosedyrer
- Gjennomgang av sykehistorien for å forstå symptomene og deres utvikling.
- Gjennomføre en nevrologisk vurdering for å evaluere muskelstyrke, reflekser og koordinasjon.
- Elektromyografi (EMG) måler muskelaktivitet og avdekker nerveproblemer.
- Nerveledningsstudier (NCS) vurderer nervefunksjon og -respons.
Utfordringer ved diagnostisering
- ALS-symptomer overlapper med andre tilstander, noe som kan føre til feildiagnostisering eller forsinket diagnose.
- Mangel på spesifikke biomarkører kan hindre tidlig og nøyaktig diagnose.
- Progresjon og symptomvariasjon gjør det utfordrende å stille diagnose.
Å skille ALS fra andre sykdommer
I motsetning til multippel sklerose (MS), som først og fremst rammer myelinskjeden i sentralnervesystemet, rammer ALS motoriske nevroner som er ansvarlige for viljestyrt muskelkontroll.
Primær lateral sklerose (PLS) har visse fellestrekk med ALS, men involverer hovedsakelig øvre motoriske nevroner, til forskjell fra ALS' kombinerte affeksjon av øvre og nedre motoriske nevroner.
I tillegg er det viktig å kunne skille mellom ALS og spinal muskelatrofi (SMA) på grunn av den felles påvirkningen på motoriske nevroner, men SMA påvirker først og fremst de nedre motoriske nevronene, i motsetning til ALS. Evnen til å skille ALS fra disse sykdommene er avgjørende for nøyaktig diagnose og skreddersydde behandlingsstrategier.
Behandling og håndtering
Behandling og håndtering av ALS omfatter en mangesidig tilnærming. Tilgjengelige behandlingsalternativer tar sikte på å lindre symptomer, bremse sykdomsutviklingen og forbedre livskvaliteten for personer med ALS.
Tilgjengelige behandlingsalternativer
- Hovedfokuset i ALS-behandlingen er å behandle symptomene, forbedre livskvaliteten og bremse sykdomsutviklingen.
- Medikamenter som riluzol og edaravone er godkjent for potensielt å forlenge overlevelsen og forsinke progresjonen.
- Baklofen og andre legemidler kan bidra til å håndtere muskelspastisitet, mens legemidler som antikolinergika kan motvirke overdreven spyttproduksjon.
- Hjelpemidler, som ikke-invasive respiratorer og ernæringssonder, hjelper til med pusting og ernæring når muskelfunksjonen avtar.
Håndtering av symptomer og livskvalitet
- Fysio- og ergoterapi bidrar til å opprettholde muskelstyrke, mobilitet og selvstendighet.
- Logopedi hjelper deg med kommunikasjons- og svelgevansker.
- Støttende behandling, som ernæringsrådgivning, psykologisk rådgivning og støttegrupper, bidrar til å løse følelsesmessige og praktiske utfordringer.
- Palliativ behandling og hospicetjenester forbedrer komfort og livskvalitet i fremskredne stadier.
Pågående forskning og fremtidige behandlinger
- Forskerne fortsetter å undersøke de underliggende årsakene til ALS og potensielle behandlingsmål.
- Målet med genterapi er å korrigere genetiske mutasjoner som er ansvarlige for familiær ALS.
- Stamcelleforskning utforsker bruk av stamceller for å erstatte skadede motoriske nevroner.
- Blant de innovative tilnærmingene er å angripe unormal proteinakkumulering og betennelse i ALS-rammede nevroner.
- Utvikling av biomarkører kan bidra til å stille tidlig diagnose og følge sykdomsutviklingen.
- Vi fortsetter å lete etter nye terapeutiske midler og strategier for å utvikle mer effektive behandlinger av ALS.
Påvirkning på dagliglivet
Å leve med ALS medfører store endringer i dagliglivet på grunn av det gradvise tapet av muskelfunksjon og motorisk kontroll. Enkle handlinger som å gå, plukke opp gjenstander og til og med snakke blir stadig vanskeligere etter hvert som sykdommen utvikler seg. Oppgaver som tidligere ble tatt for gitt, krever betydelig innsats eller kan bli umulige å utføre på egen hånd. Etter hvert som evnen til å bevege seg og kommunisere avtar, har personer med ALS ofte behov for hjelpemidler og tilpasningsteknikker for å opprettholde et visst funksjonsnivå og engasjement i de daglige rutinene.
Støtte til pasienter og pårørende
Å leve med ALS krever et robust støttenettverk for både pasienter og pårørende. Pasientene trenger fysisk, emosjonell og psykologisk støtte for å håndtere sykdommens utfordringer. Dette omfatter hjelp til daglige aktiviteter, håndtering av medisinske behov og følelsesmessig velvære.
Pårørende spiller en avgjørende rolle når det gjelder å gi denne støtten, samtidig som de søker svar på spørsmålet "Hva er amyotrofisk lateral sklerose?". De trenger også ressurser for å takle den følelsesmessige belastningen og de praktiske kravene som følger med omsorgsarbeidet. Støttegrupper, rådgivningstjenester og opplæringsressurser kan gi verdifull hjelp til å håndtere kompleksiteten ved ALS.
Fortellinger og perspektiver fra personer med ALS
Personlige historier og perspektiver fra personer som lever med ALS, gir et unikt innblikk i hvordan det er å møte denne progressive sykdommen. Disse historiene belyser styrken, motet og motstandskraften som kreves for å tilpasse seg endringene og begrensningene som ALS medfører. De gir en plattform for deling av erfaringer, utfordringer og mestringsstrategier. Deling av disse historiene skaper en følelse av fellesskap blant dem som er rammet av ALS, noe som gir håp og en følelse av samhørighet som kan hjelpe enkeltpersoner og familier til å føle seg mindre isolert.
Bli med i vårt raskt voksende fellesskap for å revolusjonere vitenskapelig kommunikasjon!
Oppdag hvordan Mind the Graph plattformen gir forskere en omfattende verktøykasse for effektiv kommunikasjon. Ved å integrere grafikk, data og informasjon på en sømløs måte forenkler Mind the Graph komplekse vitenskapelige konsepter og gjør dem tilgjengelige for både eksperter og et bredere publikum.



Abonner på nyhetsbrevet vårt
Eksklusivt innhold av høy kvalitet om effektiv visuell
kommunikasjon innen vitenskap.